It is a rare neurodegenerative disease characterized by progressive muscular dystrophy resulting in degeneration of central motor neurons found in the cortex and trunk (pyramidal cells of the motor cortex and nuclei of some motor nerves) as well as motor cells in the anterior corners of the gray matter of the spinal cord. The loss of motor neurons is associated with the degeneration of the cortical (pyramidal) path – the major motorway. Although it is a rare and still untreatable disease, it was already clinically and pathologically described in 1869 by the French neurologist Jean Martin Charcot.
In the literature, it is sometimes referred to as Charcot’s disease; in US literature, we meet with the famous American baseball player Lo Gehring – Lou Gehring’s disease.
Because it is a rare disease, incidence and prevalence are low (incidence averaging 1/50 000, prevalence averaging 1/20 000). According to the US ALS association, a comparable number of patients with ALS is diagnosed in the United States annually as with multiple sclerosis. Unlike most rare diseases, ALS affects patients aged 60 and over (80% of rare disease are affected by infants). Men have a larger predisposition (1.5: 1, males: females). Approximately two-thirds of patients with typical ALS have a spinal form that is primarily manifested in the limbs. Symptoms are focal, asymmetric muscle weakness of the lower or upper limbs. Gradually, spasticity develops atrophic limbs, responsiveness decreases, and loses the ability to hold the body properly.
Patients with ALS bulb form gradually develop dysphagia and dysarthria (speech impairment due to speech muscles impairment). The patient has primary problems with swallowing fluids, and then also with solid foods. He cannot swallow saliva, manipulate his tongue, speech, articulation and phonation is broken. Leg disability develops practically at the same time as bulbar damage, and in most patients it occurs within one to two years. Muscle weakness, fasciulism, muscle atrophy, muscle spasm, contracture to paralysis occur. The patient dies as a result of respiratory failure.
The ALS bulb form is more progressive and the average duration of the disease is 2-3 years, whereas for the ALS it is 3-5 years. Only 5% of patients survive for more than 20 years. ALS is commonly known as sporadic disease, but 5-10% of cases are family based. More than 15 genes predisposed to the disease are known today.
ALS management is supportive and palliative based, similar to other rare diseases on a multidisciplinary approach. Non-invasive ventilation prolongs life and improves its quality. The only drug with proven ability to prolong human life and improve its quality is riluzole (N07XX02 – RILUTEK, RILUZOLE ZENTIVA). According to the ATC classification, it belongs among other nervous system drugs. Riluzol does not belong to a group of orphan medicinal products, which in their development benefited from orphan status. It is, however, like many other medicines, a medicine for rare diseases. No riluzole containing drug is currently categorized in the SR. Riluzol prolongs the time until a patient needs artificial lung ventilation. The exact mechanism of riluzole interfering with the etiopathogenesis of ALS is not known. However, it is believed to reduce the excitotoxic effect of glutamate. It is likely to be a glutamate antagonist which inhibits the release of glutamate from the presynaptic terminus and an inhibitory effect on postsynaptic transmission is also contemplated. Based on the results of randomized clinical trials, riluzole prolongs life / survival by 2 months, according to uncontrolled studies, up to 20 months. Treatment of riluzole is reviewed individually by the health insurance company. The vital function of the lungs plays an important role. In advanced stages of the disease, when the patient is reliant on artificial lung ventilation, riluzole is ineffective. ALS management is mainly focused on symptomatic and supportive treatment. It often requires multidisciplinary co-operation of several specialists.
Symptomatic treatment of ALS patients includes:
amitriptyline, atropine, botulinum toxin, glandula parotis radiation – increased salivation
carbamazepine, phenytoin, gabapentin, pregabalin, diazepam, vitamin E – fasciula-
baclofen, tizanidine, diazepam – spasticity
mucolytics (carbocysteine) – dense mucosal secretion
increased fluid / fiber intake, osmotic overdosters, glycerin suppositories – constipation
diazepam – anxiety
amitriptyline, SSRI – depression
percutaneous endoscopic gastrostomy – dysphagia, weight loss
non-invasive positive pressure ventilation support, supportive ventilation mode of home care – respiratory insufficiency
morphine – dyspnea in terminal stages
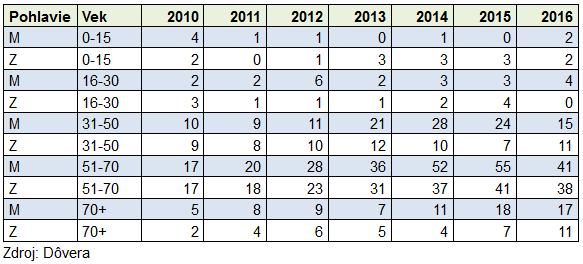
ALS is curried by a neurologist. The Center for Neuromuscular Diseases, the Neurological Clinic of the SZU and the UN in Bratislava is a specialized department. The exact number of patients with this diagnosis in Slovakia is unknown. Due to the large progression, it is estimated in tens. In 2012, the Center for neuromuscular diseases visited 17 patients with ALS (Špalek P., 2013). For these reasons, there is no separate organization in Slovakia that would only associate family members and patients with ALS. These are members in the Organization of Muscular Dystrophy in the Slovak Republic (OMD in the Slovak Republic), which we have repeatedly mentioned on the pages of the Medical Leafs.
PharmDr. Tatiana Foltánová, PhD., UK in Bratislava, Faculty of Pharmacy, Department of Pharmacology and Toxicology, Prof. MD. Ľubomír Lisý, DrSc., Department of Neurology, SZU and UNB,























 The start was planned for 9am and so after memorialize Mr. Svočák we were ready for run. The challenging track of this Half-marathon starts with decent pace. First 11 kilometers were in slightly undulated terrain. We ran around „Bagrovisko“ – a lake where I spent my summers bathing, then we passed trough a village of Batizovce and continued via beautiful meadows to village of Štôla – one of th emost beautiful places I know. A local citizens funclub was greeting us there. Afterwards my favorite walkway towards Podskalka where I heard my mommy in the distance – „Overtake a Polish!“ – which I did not understand at that time (she explained me then the Polish man was the first and she was shouting this to all participants), Juraj made me some photos and I ran further. I was slowly earning my position and really enjoyed the race when my beloved hills showed up. I mean big hills – total elevation of 650m what is more than enough for a half-marathon race. First we had a climb to „Borik“, the hill where I grew up and spent many Saturdays and Sundays with my father. At the top I was in slightly melancholic mood. Exactly at this spot we were observing the nature around, eating a snack. Oh, how wonderful childhood I had…
The start was planned for 9am and so after memorialize Mr. Svočák we were ready for run. The challenging track of this Half-marathon starts with decent pace. First 11 kilometers were in slightly undulated terrain. We ran around „Bagrovisko“ – a lake where I spent my summers bathing, then we passed trough a village of Batizovce and continued via beautiful meadows to village of Štôla – one of th emost beautiful places I know. A local citizens funclub was greeting us there. Afterwards my favorite walkway towards Podskalka where I heard my mommy in the distance – „Overtake a Polish!“ – which I did not understand at that time (she explained me then the Polish man was the first and she was shouting this to all participants), Juraj made me some photos and I ran further. I was slowly earning my position and really enjoyed the race when my beloved hills showed up. I mean big hills – total elevation of 650m what is more than enough for a half-marathon race. First we had a climb to „Borik“, the hill where I grew up and spent many Saturdays and Sundays with my father. At the top I was in slightly melancholic mood. Exactly at this spot we were observing the nature around, eating a snack. Oh, how wonderful childhood I had…